Starfysh tutorial on real dataset
Azizi Lab
Siyu He, Yinuo Jin
11-8-2022
This is a tutorial on an example real Spatial Transcriptomics (ST) data (CID44971_TNBC) from Wu et al., 2021.
Overview
Starfysh performs cell-type deconvolution followed by various downstream analyses to discover spatial interactions in tumor microenvironment. Specifically, Starfysh looks for anchor spots, defined as the spots with the highest proportion of a given cell type, and further deconvolves the remaining spots. This process is guided by user-provided signatures (see example). Starfysh provides the following options:
Base feature:
Auxiliary Variational AutoEncoder (AVAE): Spot-level deconvolution with expected cell types and corresponding annotated signature gene sets (default)
Optional:
Archetypal Analysis (AA):
If signature is not provided: (example updated soon!)
Unsupervised cell type annotation (if the input signature is not provided)
If signature is provided:
Novel cell type / cell state discovery (complementary to known cell types from the signatures)
Refine known marker genes by appending archetype-specific differentially expressed genes, and update anchor spots accordingly
Product-of-Experts (PoE) integration: Example updated soon!
Multi-modal integrative predictions with expression & histology image by leverging additional side information (e.g. cell density) from H&E image.
[1]:
import sys
IN_COLAB = "google.colab" in sys.modules
if IN_COLAB:
!pip3 install scanpy
#!pip3 install histomicstk
from google.colab import drive
drive.mount('/content/drive')
import sys
sys.path.append('/content/drive/MyDrive/SpatialModelProject/model_test_colab/')
[2]:
import os
import numpy as np
import pandas as pd
import torch
[3]:
import matplotlib.pyplot as plt
import matplotlib.font_manager
from matplotlib import rcParams
font_list = []
fpaths = matplotlib.font_manager.findSystemFonts()
for i in fpaths:
try:
f = matplotlib.font_manager.get_font(i)
font_list.append(f.family_name)
except RuntimeError:
pass
font_list = set(font_list)
plot_font = 'Helvetica' if 'Helvetica' in font_list else 'FreeSans'
rcParams['font.family'] = plot_font
rcParams.update({'font.size': 10})
rcParams.update({'figure.dpi': 300})
rcParams.update({'figure.figsize': (3,3)})
rcParams.update({'savefig.dpi': 500})
import warnings
warnings.filterwarnings('ignore')
Load starfysh
[4]:
from starfysh import (utils,
plot_utils,
starfysh,
post_analysis,
AA
)
2022-11-08 22:41:15.210400: W tensorflow/stream_executor/platform/default/dso_loader.cc:64] Could not load dynamic library 'libcudart.so.11.0'; dlerror: libcudart.so.11.0: cannot open shared object file: No such file or directory
2022-11-08 22:41:15.210419: I tensorflow/stream_executor/cuda/cudart_stub.cc:29] Ignore above cudart dlerror if you do not have a GPU set up on your machine.
(1). load data and marker genes
File Input: - Spatial transcriptomics - Count matrix: adata - (Optional): Paired histology & spot coordinates: img, map_info
Annotated signatures (marker genes) for potential cell types:
gene_sig
Starfysh is built upon scanpy and Anndata. The common ST/Visium data sample folder consists a expression count file (usually filtered_featyur_bc_matrix.h5), and a subdirectory with corresponding H&E image and spatial information, as provided by Visium platform.
For example, our example real ST data has the following structure:
├── ../data
bc_signatures_version_1013.csv
├── P1A_ER:
\__ filtered_feature_bc_mactrix.h5
├── spatial:
\__ aligned_fiducials.jpg
detected_tissue_image.jpg
scalefactors_json.json
tissue_hires_image.png
tissue_lowres_image.png
tissue_positions_list.csv
For data that doesn’t follow the common visium data structure (e.g. missing filtered_featyur_bc_matrix.h5 or the given .h5ad count matrix file lacks spatial metadata), please construct the data as Anndata synthesizing information as the example simulated data shows:
[Note]: If you’re running this tutorial locally, please download the sample data and signature gene sets, and save it in the relative path ../data (otherwise please modify the data_path defined in the cell below):
[5]:
# Load expression counts and signature gene sets
data_path = '../data/'
sample_id = 'CID44971_TNBC'
sig_name = 'bc_signatures_version_1013.csv'
adata, adata_normed = utils.load_adata(data_folder=data_path,
sample_id=sample_id, # sample id
n_genes=2000 # number of highly variable genes to keep
)
gene_sig = pd.read_csv(os.path.join(data_path, sig_name), index_col=0)
gene_sig = utils.filter_gene_sig(gene_sig, adata.to_df())
gene_sig.head()
[2022-11-08 22:41:17] Preprocessing1: delete the mt and rp
[2022-11-08 22:41:17] Preprocessing2: Normalize
[2022-11-08 22:41:17] Preprocessing3: Logarithm
[2022-11-08 22:41:17] Preprocessing4: Find the variable genes
[5]:
| LumA | LumB | MBC | CSC | Normal epithelial | Tcm | Tem | Tfh | Treg | Activated CD8 | ... | Plasmablasts | MDSC | Monocytes | cDC | pDC | CAFs MSC iCAF-like | CAFs myCAF-like | PVL differentiated | PVL immature | Endothelial | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basal | |||||||||||||||||||||
| EMP1 | SH3BGRL | UGCG | COL11A2 | CD44 | KRT14 | CCR7 | IL7R | CXCL13 | TNFRSF4 | CD69 | ... | IGKV3-15 | ITGAM | LYZ | CD80 | IL3RA | APOD | COL1A1 | ACTA2 | CCL19 | ACKR1 |
| TAGLN | HSPB1 | ARMT1 | SDC1 | ESA | KRT17 | LTB | ANXA1 | NMB | LTB | CCR7 | ... | IGHG1 | CD33 | IL1B | CD86 | LILRA4 | DCN | COL1A2 | TAGLN | RGS5 | FABP4 |
| TTYH1 | PHGR1 | ISOC1 | FBN2 | CD133 | LTF | IL7R | CXCR4 | NR3C1 | IL32 | CD27 | ... | IGKV1-5 | ARG1 | G0S2 | CCR7 | CD123 | PTGDS | COL3A1 | MYL9 | IGFBP7 | PLVAP |
| RTN4 | SOX9 | GDF15 | MMP1 | ALDH1 | KRT15 | SARAF | KLRB1 | DUSP4 | BATF | BTLA | ... | IGKV3-20 | NOS2 | TYROBP | CD1A | TCF4 | CFD | LUM | TPM2 | NDUFA4L2 | RAMP2 |
| TK1 | CEBPD | ZFP36 | FABP5 | CD24 | PTN | SELL | TNFAIP3 | TNFRSF18 | FOXP3 | CD40LG | ... | IGKV3-11 | CD68 | FCN1 | CD1C | IRF7 | LUM | SFRP2 | NDUFA4L2 | CCL2 | VWF |
5 rows × 29 columns
[6]:
adata
[6]:
AnnData object with n_obs × n_vars = 1162 × 2000
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'sample', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt'
var: '_index', 'features', 'highly_variable'
[7]:
# Load spatial information
img, map_info = utils.preprocess_img(data_path,
sample_id,
adata_index=adata.obs.index,
hchannal=False
)
umap_df = utils.get_umap(adata, display=True)
... storing 'sample' as categorical
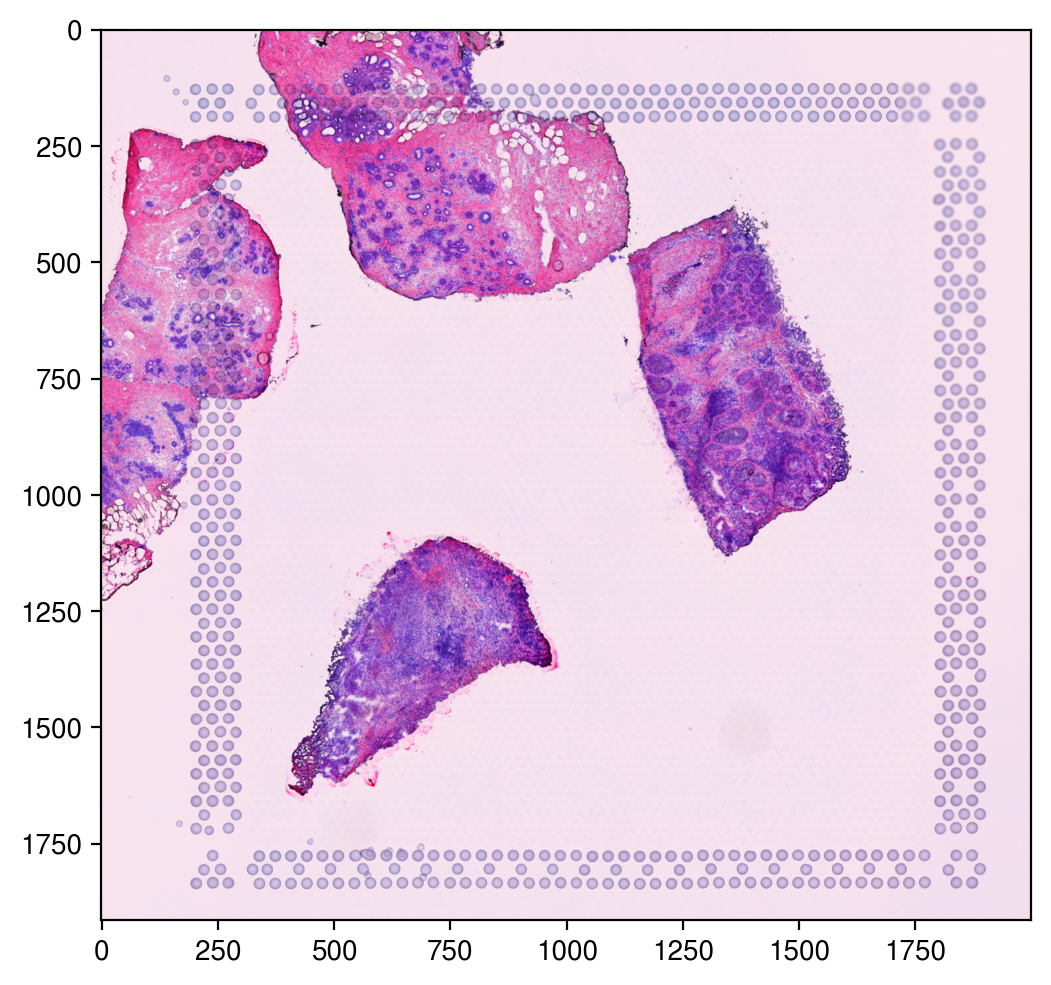
[8]:
plt.figure(figsize=(6, 6), dpi=200)
plt.imshow(img)
plt.show()
[9]:
map_info.head()
[9]:
| array_row | array_col | imagerow | imagecol | sample | |
|---|---|---|---|---|---|
| AACATTGGTCAGCCGT-1 | 3 | 17 | 282.131013 | 520.522390 | CID44971_TNBC |
| CATCGAATGGATCTCT-1 | 3 | 19 | 282.131013 | 543.117747 | CID44971_TNBC |
| CGGGTTGTAGCTTTGG-1 | 3 | 21 | 282.131013 | 565.920401 | CID44971_TNBC |
| CCTAAGTGTCTAACCG-1 | 2 | 22 | 262.437812 | 577.114430 | CID44971_TNBC |
| TCTGTGACTGACCGTT-1 | 3 | 23 | 282.131013 | 588.515757 | CID44971_TNBC |
(2). Preprocessing (finding anchor spots)
Identify anchor spot locations
Instantiate parameters for Starfysh model training: - Raw & normalized counts after taking highly variable genes - filtered signature genes - library size & spatial smoothed library size (log-transformed) - Anchor spot indices (anchors_df) for each cell type & their signature means (sig_means)
[10]:
# Parameters for training
visium_args = utils.VisiumArguments(adata,
adata_normed,
gene_sig,
map_info,
n_anchors=60,
window_size=3
)
adata, adata_normed = visium_args.get_adata()
[2022-11-08 22:41:30] Filtering signatures not highly variable...
[2022-11-08 22:41:30] Smoothing library size by taking averaging with neighbor spots...
The number of original variable genes in the dataset (2000,)
The number of siganture genes in the dataset (781,)
After filter out some genes in the signature not in the var_names ... (194,)
After filter out some genes not highly expressed in the signature ... (194,)
Combine the varibale and siganture, the total unique gene number is ... 2000
The number of original variable genes in the dataset (2000,)
The number of siganture genes in the dataset (781,)
After filter out some genes in the signature not in the var_names ... (0,)
After filter out some genes not highly expressed in the signature ... (0,)
Combine the varibale and siganture, the total unique gene number is ... 2000
[2022-11-08 22:41:31] Retrieving & normalizing signature gene expressions...
[2022-11-08 22:41:31] Identifying anchor spots (highly expression of specific cell-type signatures)...
Visualize the spatial data
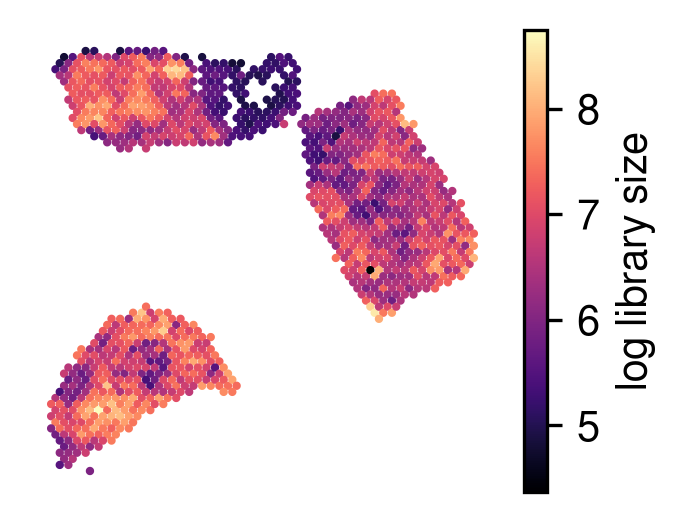
[11]:
plot_utils.plot_spatial_feature(adata,
map_info,
visium_args.log_lib,
label='log library size'
)
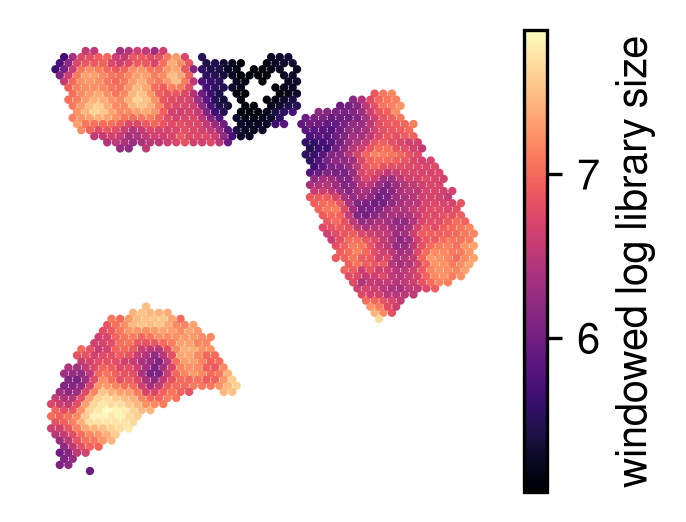
[12]:
plot_utils.plot_spatial_feature(adata,
map_info,
visium_args.win_loglib,
label='windowed log library size',
)
plot raw gene expression:
[19]:
plot_utils.plot_spatial_gene(adata,
map_info,
gene_name='IL7R')
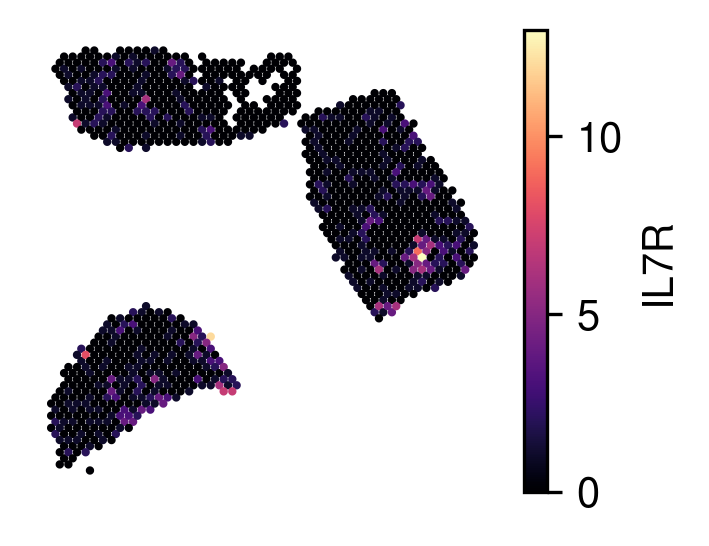
Visualize anchor spots
[20]:
plot_utils.plot_anchor_spots(umap_df,
visium_args.pure_spots,
visium_args.sig_mean,
bbox_x=2
)

(3). Optional: appending anchor spots with archetypal analysis
Overview: If users don’t provide annotated gene signature sets with cell types, Starfysh annotates cell types via archetypal analysis (AA). The underlying assumption is that the geometric “extremes” are identified as the purest cell types, whereas all other spots are mixture of the “archetypes”. If the users provide the gene signature sets, they can still optionally apply AA to refine marker genes and update anchor spots for known cell types. In addition, AA can identify potential novel cell types / states. Here are the overview features provided by the AA step: - Finding archetypal spots & assign 1-1 mapping to their closest anchor spot neighbors - Finding archetypal marker genes & append them to marker genes of annotated cell types - Find novel cell type / cell states as the most distant archetypes
[21]:
aa_model = AA.ArchetypalAnalysis(adata_orig=adata_normed)
archetype, arche_dict, major_idx, evs = aa_model.compute_archetypes(r=40)
# (1). Find archetypal spots & archetypal clusters
arche_df = aa_model.find_archetypal_spots(major=True)
# (2). Find marker genes associated with each archetypal cluster
markers_df = aa_model.find_markers(n_markers=30, display=False)
# (3). Map archetypes to closest anchors (1-1 per cell type)
anchors_df = visium_args.get_anchors()
map_df, map_dict = aa_model.assign_archetypes(anchors_df)
# (4). Optional: Find the most distant archetypes that are not assigned to any annotated cell types
distant_arches = aa_model.find_distant_archetypes(anchors_df, n=3)
[2022-11-08 22:43:09] Computing intrinsic dimension to estimate k...
30 components are retained using conditional_number=30.00
[2022-11-08 22:43:10] Estimating lower bound of # archetype as 12...
[2022-11-08 22:43:23] Calculating UMAPs for counts + Archetypes...
[2022-11-08 22:43:27] Calculating UMAPs for counts + Archetypes...
[2022-11-08 22:43:29] 0.6918 variance explained by raw archetypes.
Merging raw archetypes within 40 NNs to get major archetypes
[2022-11-08 22:43:29] Finding 20 nearest neighbors for each archetype...
[2022-11-08 22:43:29] Finding 30 top marker genes for each archetype...
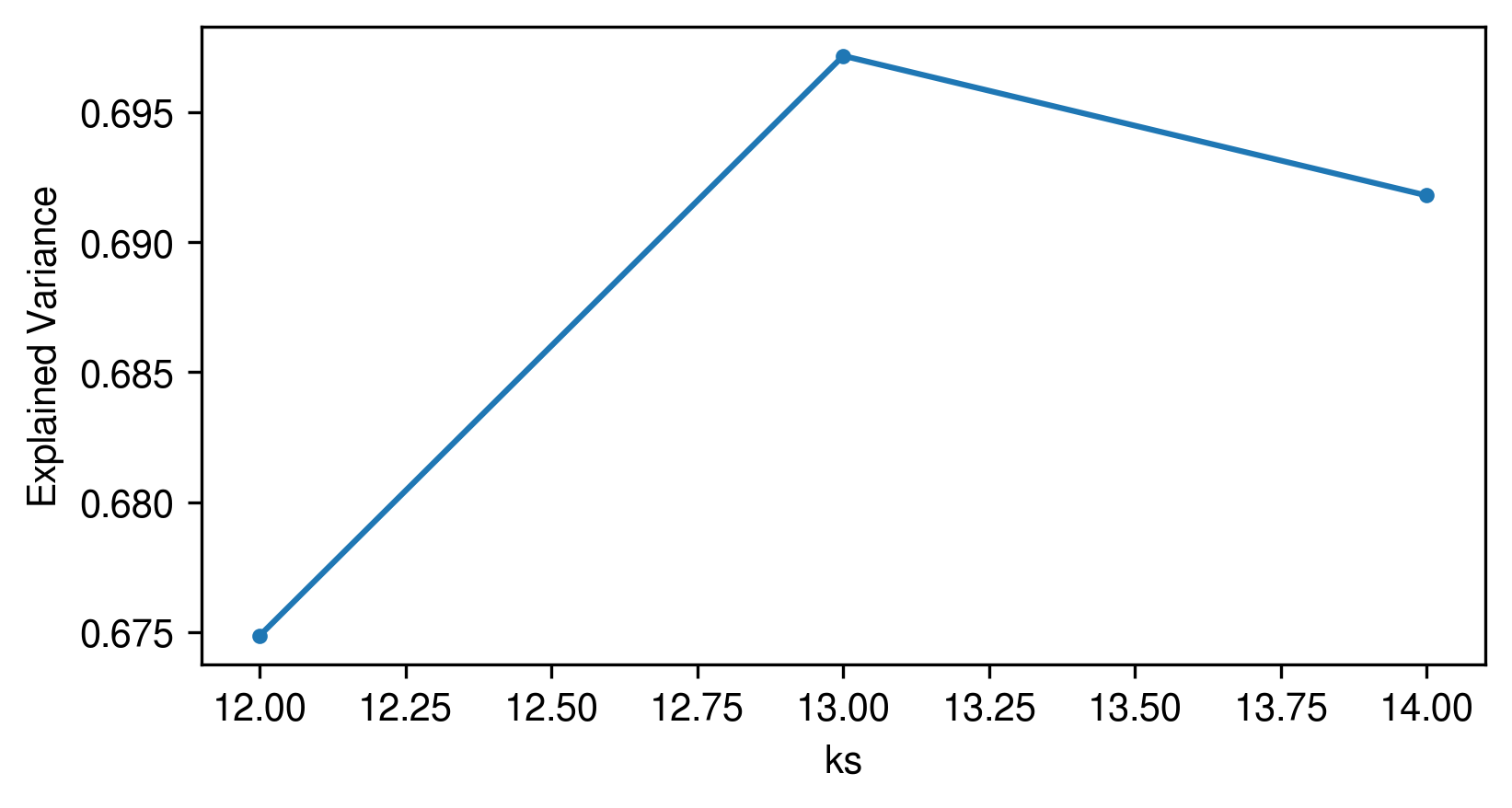
[22]:
plot_utils.plot_evs(evs, kmin=aa_model.kmin)
Visualize archetypes
[25]:
aa_model.plot_archetypes(do_3d=False, major=True, disp_cluster=False)
[2022-11-08 22:44:18] No handles with labels found to put in legend.
[25]:
(<Figure size 1800x1200 with 1 Axes>, <AxesSubplot:>)

Visualize overlapping ratio between archetypal & anchor spots:
[26]:
aa_model.plot_mapping(map_df)
[26]:
<seaborn.matrix.ClusterGrid at 0x7f3366e01910>
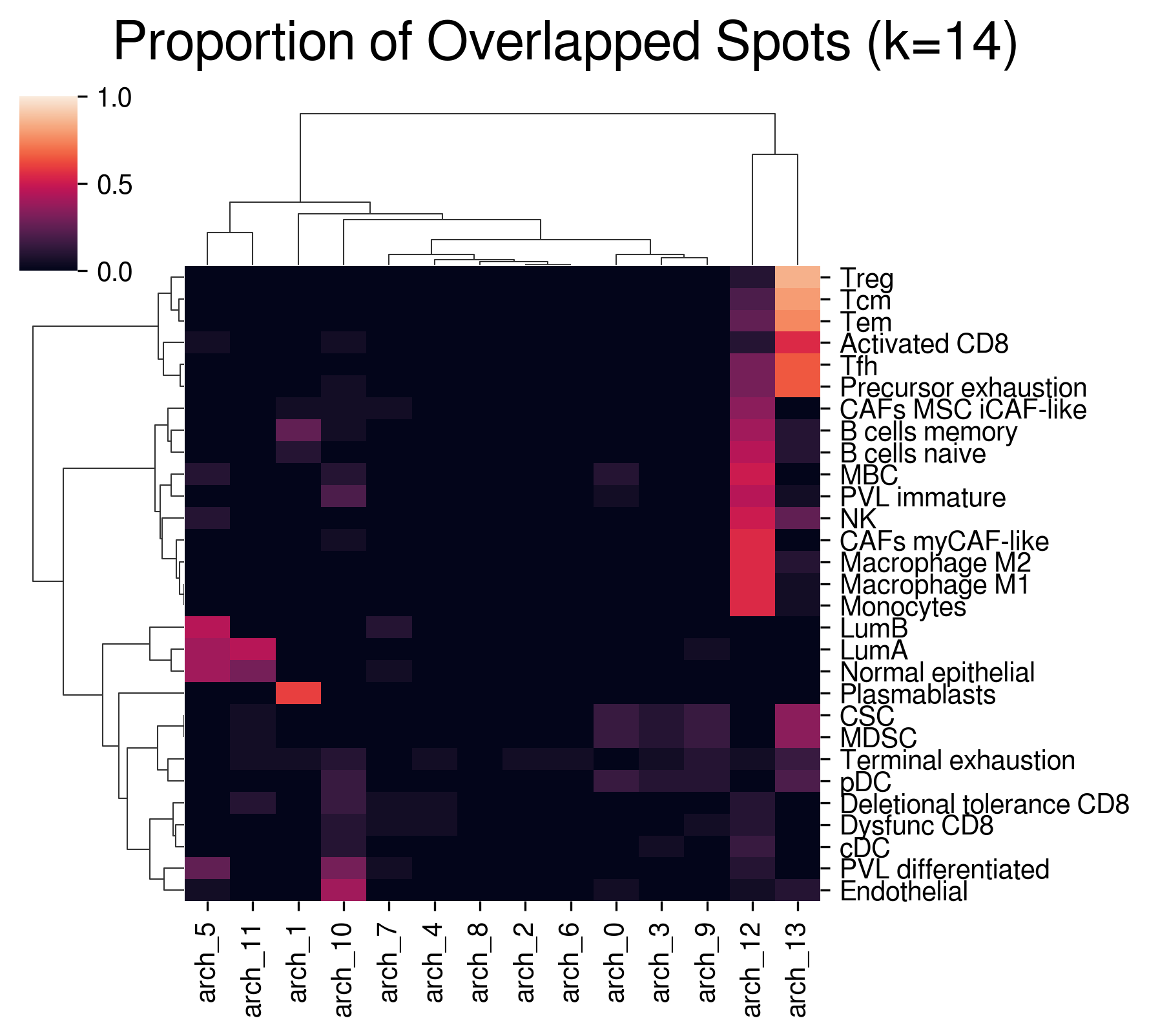
Application: appending marker genes Append archetypal marker genes with the best-aligned anchors:
[27]:
visium_args = utils.refine_anchors(
visium_args,
aa_model,
map_info,
n_genes=5,
n_iters=1
)
[2022-11-08 22:44:56] Finding 50 top marker genes for each archetype...
[2022-11-08 22:44:57] Filtering signatures not highly variable...
[2022-11-08 22:44:57] Smoothing library size by taking averaging with neighbor spots...
Refining round 1...
appending 5 genes in arch_5 to LumA...
appending 5 genes in arch_11 to LumA...
appending 5 genes in arch_5 to LumB...
appending 5 genes in arch_12 to MBC...
appending 5 genes in arch_13 to CSC...
appending 5 genes in arch_5 to Normal epithelial...
appending 5 genes in arch_13 to Tcm...
appending 5 genes in arch_13 to Tem...
appending 5 genes in arch_13 to Tfh...
appending 5 genes in arch_13 to Treg...
appending 5 genes in arch_13 to Activated CD8...
appending 5 genes in arch_13 to Precursor exhaustion...
appending 5 genes in arch_12 to NK...
appending 5 genes in arch_12 to B cells memory...
appending 5 genes in arch_12 to B cells naive...
appending 5 genes in arch_12 to Macrophage M1...
appending 5 genes in arch_12 to Macrophage M2...
appending 5 genes in arch_1 to Plasmablasts...
appending 5 genes in arch_13 to MDSC...
appending 5 genes in arch_12 to Monocytes...
appending 5 genes in arch_12 to CAFs MSC iCAF-like...
appending 5 genes in arch_12 to CAFs myCAF-like...
appending 5 genes in arch_12 to PVL immature...
appending 5 genes in arch_10 to Endothelial...
The number of original variable genes in the dataset (2000,)
The number of siganture genes in the dataset (811,)
After filter out some genes in the signature not in the var_names ... (194,)
After filter out some genes not highly expressed in the signature ... (194,)
Combine the varibale and siganture, the total unique gene number is ... 2000
The number of original variable genes in the dataset (2000,)
The number of siganture genes in the dataset (811,)
After filter out some genes in the signature not in the var_names ... (30,)
After filter out some genes not highly expressed in the signature ... (30,)
Combine the varibale and siganture, the total unique gene number is ... 2000
[2022-11-08 22:44:58] Retrieving & normalizing signature gene expressions...
[2022-11-08 22:44:58] Identifying anchor spots (highly expression of specific cell-type signatures)...
Run starfysh
We perform n_repeat random restarts and select the best model with lowest loss for parameter c (inferred cell-type proportions):
(1). Model parameters
[28]:
n_repeats = 3
epochs = 100
patience = 10
device = torch.device('cuda')
(2). Model training
Base Starfysh deconvolution with ST count matrix
[29]:
# Run models
model, loss = utils.run_starfysh(visium_args,
n_repeats=n_repeats,
epochs=epochs,
patience=patience,
device=device
)
[2022-11-08 22:45:22] Running Starfysh with 3 restarts, choose the model with best parameters...
[2022-11-08 22:45:22] === Restart Starfysh 1 ===
[2022-11-08 22:45:24] Initializing model parameters...
[2022-11-08 22:45:32] Epoch[10/100], train_loss: 2331.6271, train_reconst: 980.7252, train_z: 17.5547,train_c: 1303.9635,train_n: 29.3837
[2022-11-08 22:45:40] Epoch[20/100], train_loss: 2057.6486, train_reconst: 953.3014, train_z: 17.2354,train_c: 1060.3301,train_n: 26.7817
[2022-11-08 22:45:48] Epoch[30/100], train_loss: 1874.9804, train_reconst: 928.6902, train_z: 17.4447,train_c: 902.8640,train_n: 25.9815
[2022-11-08 22:45:56] Epoch[40/100], train_loss: 1776.9664, train_reconst: 917.4840, train_z: 18.1390,train_c: 816.0717,train_n: 25.2718
[2022-11-08 22:46:04] Epoch[50/100], train_loss: 1771.6806, train_reconst: 901.6704, train_z: 18.5714,train_c: 826.2227,train_n: 25.2161
[2022-11-08 22:46:12] Epoch[60/100], train_loss: 1641.5248, train_reconst: 890.0628, train_z: 19.4926,train_c: 707.0521,train_n: 24.9172
[2022-11-08 22:46:19] Epoch[70/100], train_loss: 1636.3772, train_reconst: 881.5967, train_z: 20.1486,train_c: 709.6434,train_n: 24.9885
[2022-11-08 22:46:27] Epoch[80/100], train_loss: 1541.4552, train_reconst: 872.4099, train_z: 20.7251,train_c: 623.5006,train_n: 24.8195
[2022-11-08 22:46:35] Epoch[90/100], train_loss: 1520.7062, train_reconst: 865.3784, train_z: 21.4300,train_c: 609.1164,train_n: 24.7815
[2022-11-08 22:46:43] Epoch[100/100], train_loss: 1486.8839, train_reconst: 854.5322, train_z: 22.0613,train_c: 585.7725,train_n: 24.5179
[2022-11-08 22:46:43] === Finished training ===
[2022-11-08 22:46:43] === Restart Starfysh 2 ===
[2022-11-08 22:46:43] Initializing model parameters...
[2022-11-08 22:46:51] Epoch[10/100], train_loss: 2179.7421, train_reconst: 984.0697, train_z: 17.0742,train_c: 1148.4680,train_n: 30.1303
[2022-11-08 22:46:59] Epoch[20/100], train_loss: 1915.0692, train_reconst: 954.3004, train_z: 16.6733,train_c: 917.3649,train_n: 26.7305
[2022-11-08 22:47:07] Epoch[30/100], train_loss: 1829.6925, train_reconst: 938.8659, train_z: 17.5139,train_c: 847.2063,train_n: 26.1064
[2022-11-08 22:47:16] Epoch[40/100], train_loss: 1737.7362, train_reconst: 919.4831, train_z: 18.0926,train_c: 774.6181,train_n: 25.5424
[2022-11-08 22:47:24] Epoch[50/100], train_loss: 1649.1499, train_reconst: 908.0915, train_z: 18.4772,train_c: 697.1203,train_n: 25.4610
[2022-11-08 22:47:32] Epoch[60/100], train_loss: 1612.0537, train_reconst: 894.6327, train_z: 18.9822,train_c: 673.2488,train_n: 25.1900
[2022-11-08 22:47:40] Epoch[70/100], train_loss: 1549.7182, train_reconst: 880.9087, train_z: 20.0021,train_c: 623.9455,train_n: 24.8620
[2022-11-08 22:47:48] Epoch[80/100], train_loss: 1569.1090, train_reconst: 874.8611, train_z: 20.5701,train_c: 648.8480,train_n: 24.8299
[2022-11-08 22:47:48] Saving best-performing model; Early stopping...
[2022-11-08 22:47:48] === Finished training ===
[2022-11-08 22:47:48] === Restart Starfysh 3 ===
[2022-11-08 22:47:48] Initializing model parameters...
[2022-11-08 22:47:56] Epoch[10/100], train_loss: 2466.4759, train_reconst: 979.0706, train_z: 18.0902,train_c: 1440.5294,train_n: 28.7857
[2022-11-08 22:48:04] Epoch[20/100], train_loss: 2115.0540, train_reconst: 954.9546, train_z: 17.2289,train_c: 1116.2076,train_n: 26.6629
[2022-11-08 22:48:12] Epoch[30/100], train_loss: 1915.3935, train_reconst: 936.5548, train_z: 17.6840,train_c: 935.5861,train_n: 25.5686
[2022-11-08 22:48:20] Epoch[40/100], train_loss: 1810.5590, train_reconst: 917.5890, train_z: 18.1688,train_c: 849.7304,train_n: 25.0707
[2022-11-08 22:48:29] Epoch[50/100], train_loss: 1755.5792, train_reconst: 901.9857, train_z: 18.8673,train_c: 809.9228,train_n: 24.8033
[2022-11-08 22:48:37] Epoch[60/100], train_loss: 1693.6489, train_reconst: 889.8988, train_z: 19.5530,train_c: 759.4232,train_n: 24.7739
[2022-11-08 22:48:46] Epoch[70/100], train_loss: 1605.5453, train_reconst: 883.8405, train_z: 19.7191,train_c: 677.4439,train_n: 24.5418
[2022-11-08 22:48:54] Epoch[80/100], train_loss: 1607.9693, train_reconst: 875.2010, train_z: 20.5664,train_c: 687.6567,train_n: 24.5451
[2022-11-08 22:49:00] Epoch[88/100], train_loss: 1620.8909, train_reconst: 864.1657, train_z: 21.0883,train_c: 711.0296,train_n: 24.6073
[2022-11-08 22:49:00] Saving best-performing model; Early stopping...
[2022-11-08 22:49:00] === Finished training ===
Starfysh deconvolution with histology integration (PoE)
Available soon!
(3). Downstream analysis
Extract Starfysh inference
[41]:
inference_outputs, generative_outputs, px = starfysh.model_eval(model,
adata,
visium_args.sig_mean,
device,
visium_args.log_lib,
)
u = post_analysis.get_z_umap(inference_outputs, map_info)
u = pd.DataFrame(u,columns=['umap1','umap2'])
u.index = map_info.index
Visualize starfysh deconvolution results
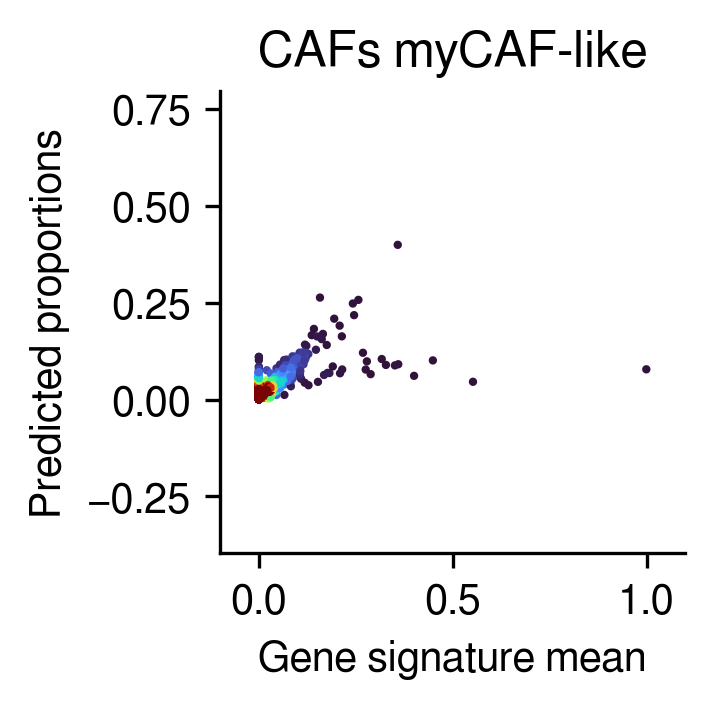
Gene sig mean vs. inferred prop
[31]:
n_cell_types = gene_sig.shape[1]
idx = np.random.randint(0, n_cell_types)
post_analysis.gene_mean_vs_inferred_prop(inference_outputs,
visium_args,
idx=idx
)
Spatial visualizations:
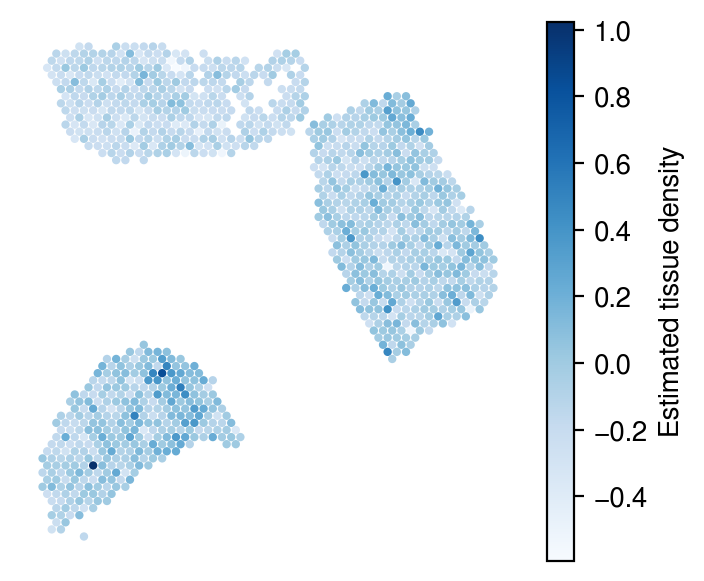
Inferred density
[32]:
plot_utils.pl_spatial_inf_feature(adata,
map_info,
inference_outputs,
feature='ql_m',
idx=0,
plt_title='',
label='Estimated tissue density',
s=4,
cmap='Blues'
#vmax=3
)
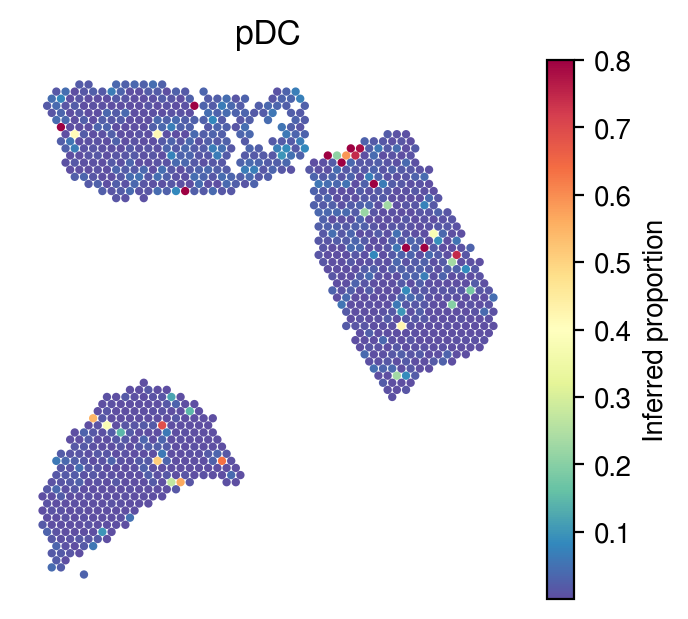
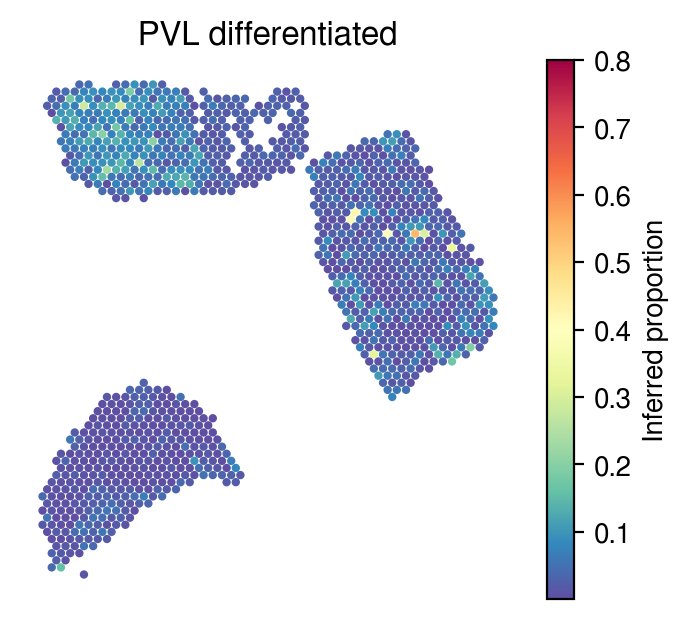
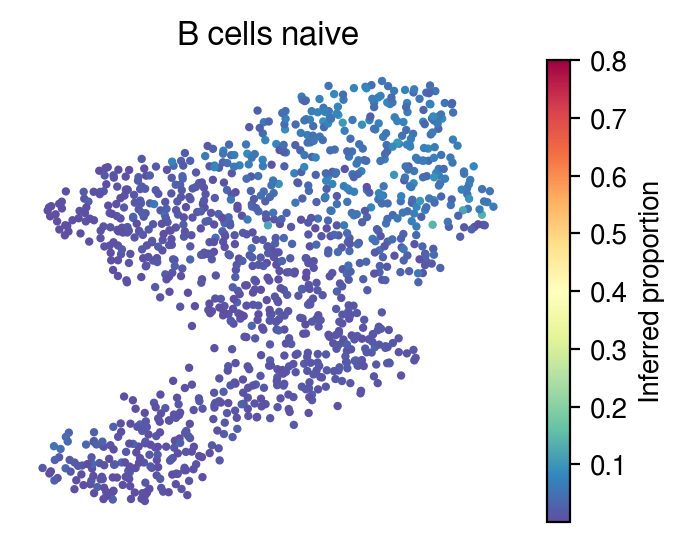
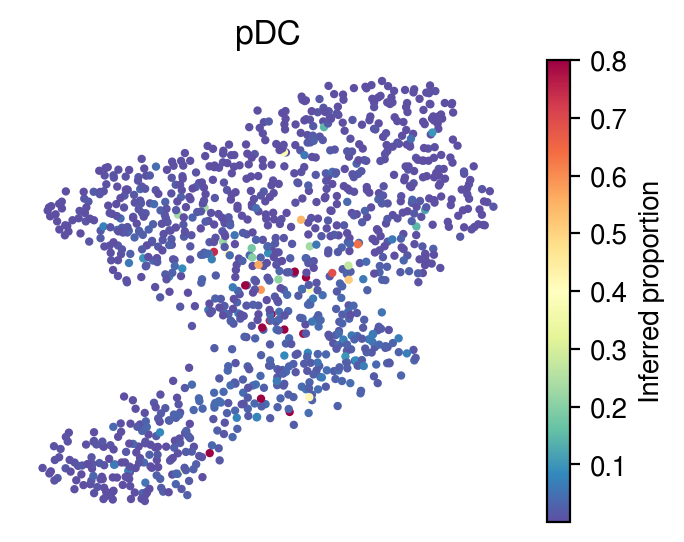
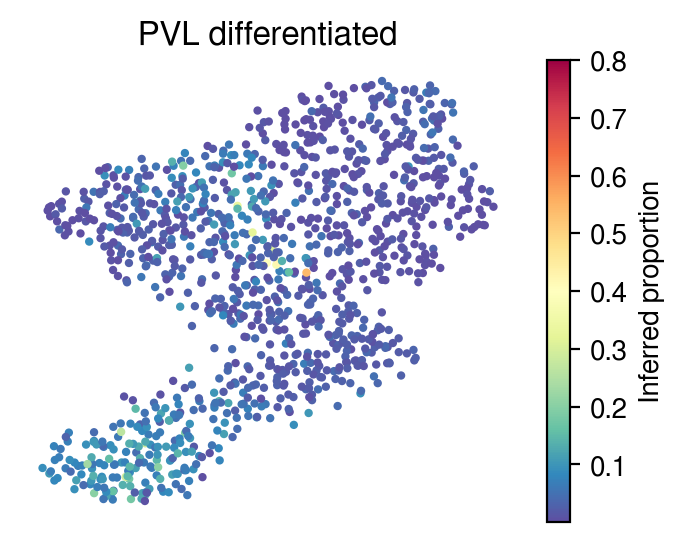
Inferred proportions
Choose random cell types
[34]:
celltype_indices = np.random.randint(0, n_cell_types, size=5)
[35]:
for idx in celltype_indices:
# Inferred proportions on ST spatial coordinates
plot_utils.pl_spatial_inf_feature(adata,
map_info,
inference_outputs,
feature='qc_m',
idx=idx,
plt_title=gene_sig.columns[idx],
label='Inferred proportion',
s=4,
vmax=0.8
)
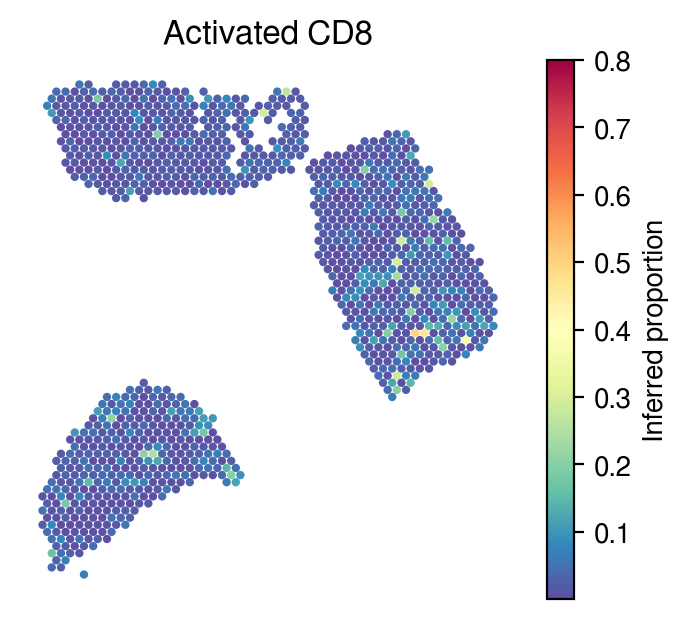
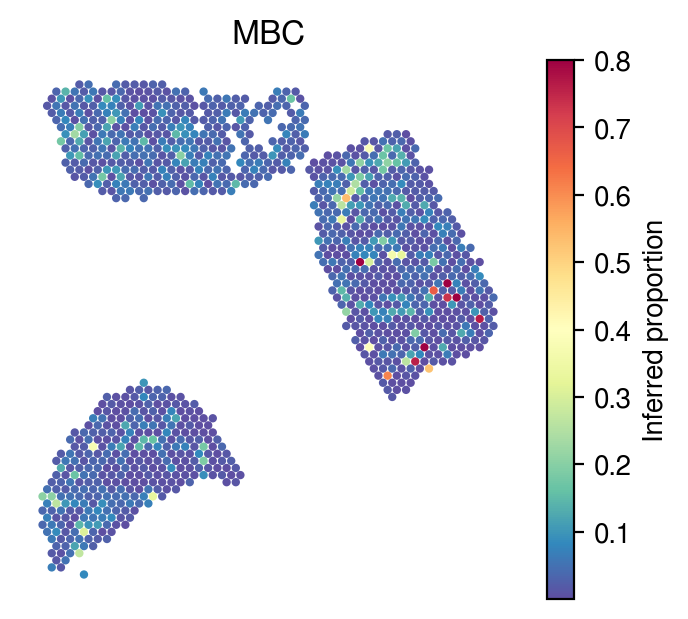
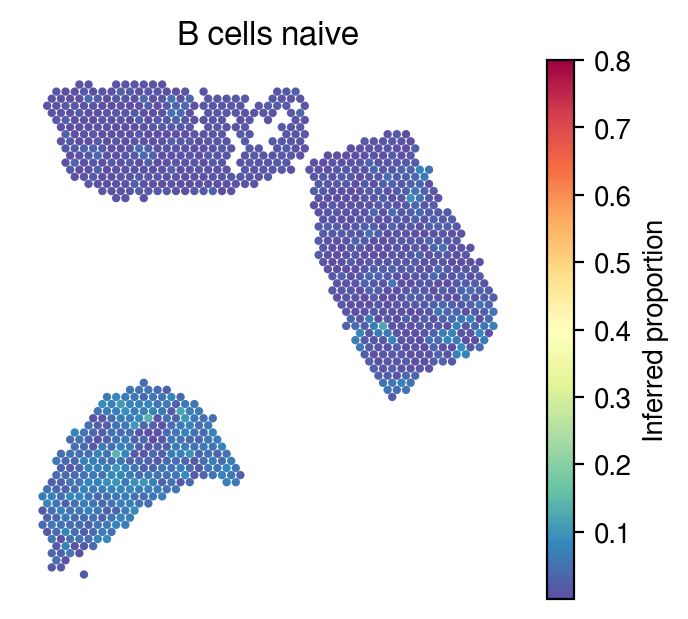
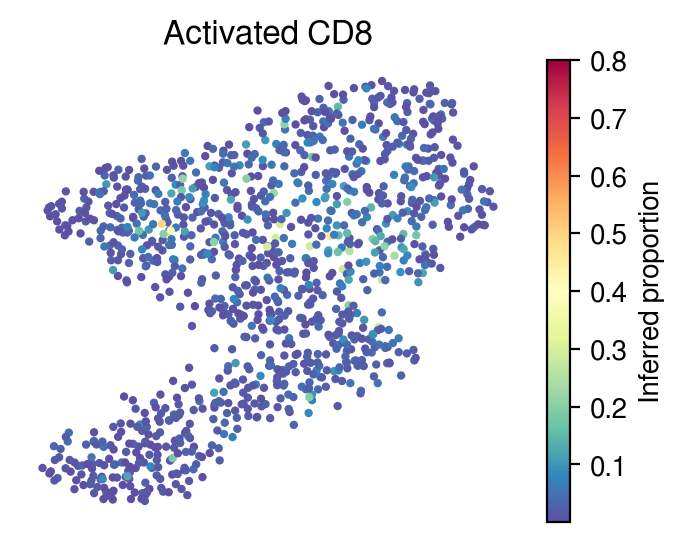
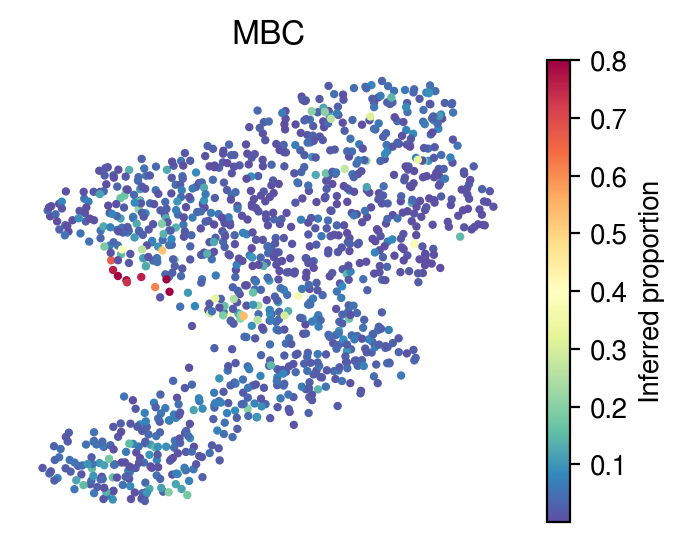
[36]:
for idx in celltype_indices:
# Inferred proportions on starfysh learnt latent dimension
plot_utils.pl_umap_feature(adata,
u,
inference_outputs,
feature='qc_m',
idx=idx,
plt_title=gene_sig.columns[idx],
label='Inferred proportion',
s=4,
vmax=0.8)
Infer cell-type specific expressions for each spot
[37]:
pred_exp = {}
for i, label in enumerate(gene_sig.columns):
pred_exp[label] = starfysh.model_ct_exp(model,
adata,
visium_args.sig_mean_znorm,
device,
visium_args.log_lib,
ct_idx=i
).tolist()
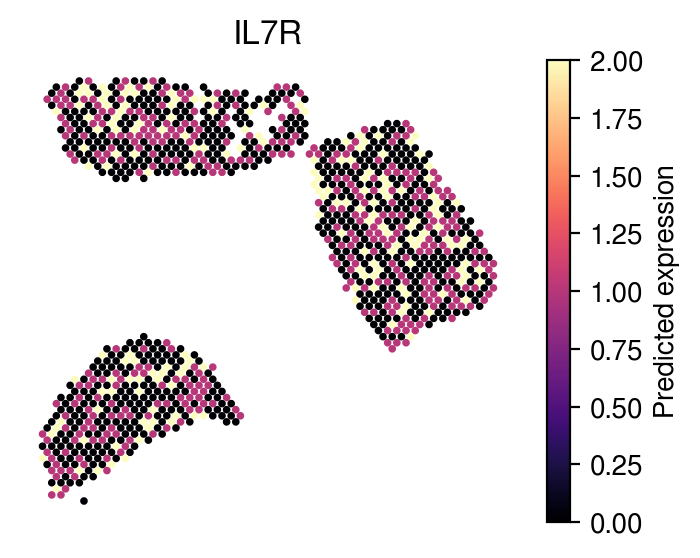
Plot spot-level expression of IL7R from Effector Memory T cells (Tem):
[40]:
sample_gene = 'IL7R'
sample_cell_type = 'Tem'
idx = list(adata.var_names[adata.var['highly_variable']]).index(sample_gene)
plot_utils.pl_spatial_inf_gene(adata,
map_info,
feature=np.asarray(pred_exp[sample_cell_type]),
idx= idx,
plt_title=list(adata.var_names[adata.var['highly_variable']])[idx],
label='Predicted expression',
s=3,
vmax=2)
[ ]: